Методы определения порядка реакции
На практике исследование скорости химических реакций начинают с определения порядков этой реакции по каждому из реагирующих веществ и (или) порядка реакции в целом. Для реакции с участием нескольких веществ определить 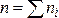 можно только поэтапно, определяя порядки по отдельным веществам. Для этого используется метод избытка (метод изоляции, метод Оствальда), а затем определяют ni любым из описанных ниже методов.
можно только поэтапно, определяя порядки по отдельным веществам. Для этого используется метод избытка (метод изоляции, метод Оствальда), а затем определяют ni любым из описанных ниже методов.
Метод изоляции или метод избытка.
Если в исследуемой реакции участвуют несколько реагирующих веществ, то общий порядок реакции , где ni - порядок по каждому из участников. Частный порядок ni можно определить, взяв все вещества, кроме данного, взять в большом избытке. Так, в случае реакции
А + В + С ® D
скорость равна
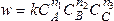 .
.
Если вещества В и С взяты в большом избытке, то при протекании реакции их концентрации практически не изменяются, т.е.
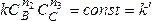 .
.
И
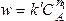 .
.
Тогда, определив порядки реакции по каждому из реагирующих веществ, можно найти общий порядок реакции:
n = n1 + n2 + n3.
Метод подстановки.
Этот метод заключается в том, что опытные результаты - текущие концентрации веществ в моменты времени t - последовательно подставляются в интегральные кинетические уравнения реакций нулевого (7), первого (9), второго (12 или 14) порядков и определяется, какое из уравнений дает практически постоянную величину константы скорости. Именно это уравнение и определяет порядок исследуемой реакции.
Графический вариант метода подстановки.
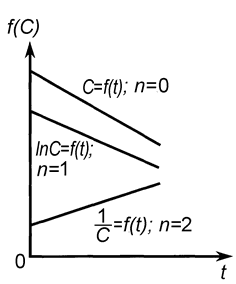
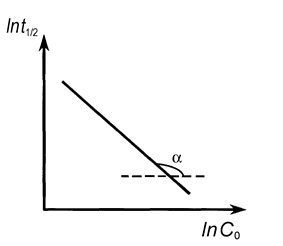
| Рис. 9. Линейные зависимости различных функций концентрации реагирующих веществ от времени. | Рис. 10. Определение порядка реакции по зависимости времени полупревращения от начальной концентрации |
Графический метод состоит в построении зависимостей различных функций концентрации от времени и определения, для какой из них наблюдается прямолинейная зависимость.
Линейные зависимости для реакций различных порядков получаются в следующих координатах (рис. 9): нулевого порядка С = f(t), первого порядка lnС = f(t), второго порядка 1/С = f(t).
Для реакций более высоких порядков выбираются координаты  -t.
-t.
Метод определения порядка реакции по времени полупревращения.
Порядок реакции определяется на основе опытной зависимости времен полупревращения (t1/2) от начальных концентраций веществ (C0).
Как уже отмечалось, для n = 0 время полупревращения прямо пропорционально величине С0, для n = 1 - не зависит от С0, а n = 2 - обратно пропорционально С0. Для реакции любого другого порядка справедливо соотношение (20)
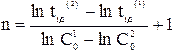 , (20)
, (20)
где t1/2(1) и t1/2(2) - время полупревращения при начальных концентрациях С01 и С02, соответственно.
Возможен графический вариант метода: по оси ординат откладываются значения ln(t1/2), а по оси абсцисс - значения lnC0 (рис. 10), тогда n = -tga + 1.
Дифференциальный метод Вант-Гоффа.
Этот метод применяют, когда в реакции участвует одно вещество, или при участии нескольких веществ, когда все вещества, кроме данного (А), взяты в большом избытке. Тогда скорость реакции равна
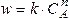 . (21)
. (21)
Прологарифмируем уравнение (21):
lnw = lnk + n lnС
Очевидно, что в координатах lnw- lnC мы получим линейную зависимость, причем n = tga.
Построив зависимость lnw от lnC (рис. 11), можно определить порядок реакции, равный в данном случае тангенсу угла наклона прямой (n = tga).
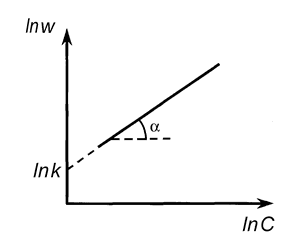
Рис. 11. Определение порядка реакции дифференциальным методом Вант-Гоффа.
studopedia.ru
20.4. Методы определения порядка реакции
При определении порядка реакции вначале находят порядок по каждому из реагирующих веществ. Для этого концентрации всех веществ, кроме рассматриваемого, берутся в большом избытке, так, что их можно считать постоянными и ввести в константу скорости. Используя какие-либо методы анализа, определяют концентрации исследуемого вещества через различные промежутки времени. Для того, чтобы концентрация вещества не изменилась во время взятия пробы и проведения анализа, реакцию затормаживают (“замораживают”) – охлаждают реакционную смесь, вводят специальные реактивы и т.п. Существует много различных способов определения порядка, наиболее распространенные из которых мы рассмотрим.
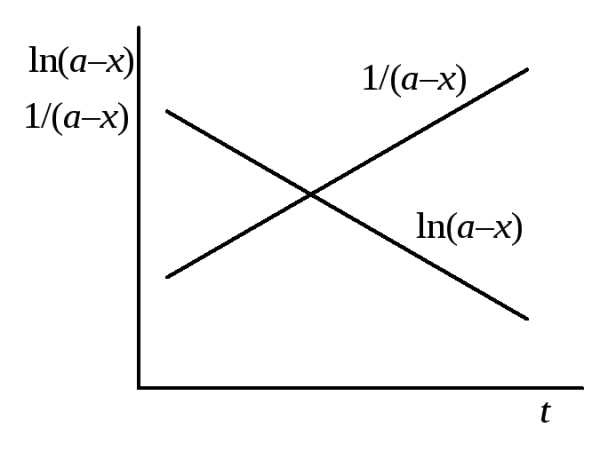
Рис.20.3. Определение порядка реакции методом графического подбора
1.Метод графического подбора. Как следует из уравнения (20.10) для реакции первого порядка выполняется линейная зависимость в координатах логарифм концентрации – время. Для реакций второго порядка такая зависимость наблюдается в координатах 1/(a – x) –t(уравнение (20.17)), а для реакцийn-ого порядка прямая получается в координатах 1/(a–x) – время (уравнение (20.30)). Таким образом, используя полученные в эксперименте значения концентраций в различные моменты времени, строят графики в тех или координатах до получения линейной зависимости.2. Метод аналитического подбора уравнениязаключается в том, что проводится расчет константы скорости путем подстановки экспериментальных данных в различные кинетические уравнения. Если уравнение выбрано правильно, константа скорости должна оставаться постоянной в пределах ошибок опыта не зависимо от времени; систематический ход константы скорости свидетельствует о том, что уравнение выбрано неверно.
3. Определение порядка по периоду полуреакции. Логарифмируя уравнение (20.31) для периода полуреакции, получим:
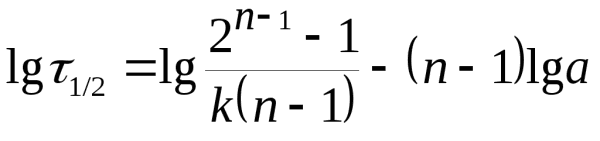 . (20.33)
. (20.33)
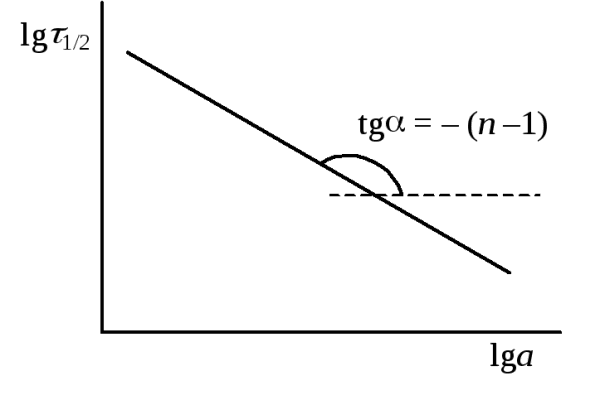
Рис.20.4. Определение порядка по периоду полуреакции
Это линейная зависимость в координатах lg1/2– lga. Построив график в этих координатах (рис. 20.3) по тангенсу угла наклона прямой определяем порядок реакции. Отрезок, отсекаемый прямой на оси ординат равен4. Графический метод определения порядка. Скорость реакцииn-ого порядка по данному веществу равна
v = kcn или
lgv= lgk+nlgc, (20.34)
где c– текущая концентрация реагирующего вещества.
Для определения порядка вначале строят график зависимости концентрация – время. Проводя касательные к кривой в точках, соответствующим различным моментам времени t1,t2, …, находят по тангенсу угла наклона касательных скорости реакцииv1,v2, … в эти моменты времени (рис. 20.5a). Затем логарифмы скоростей откладывают как функции логарифмов соответствующих концентрацийc1,c2,.... Согласно уравнению (20.25) должна получиться прямая, тангенс угла наклона которой равен порядку реакции, а отсекаемый на оси ординат отрезок – логарифму константы скорости (рис. 20.5б).
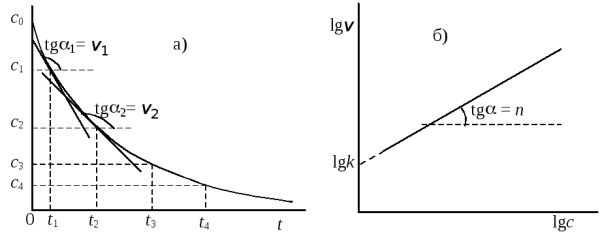
Рис.20.5. Определение порядка реакции графическим методом:
a) нахождение скорости; б) нахождение порядка и константы скорости
Существуют также другие методы определения порядка реакций. Для надежного определения нахождения этой величины обычно необходимо использовать несколько методов.
StudFiles.ru
Методы определения порядка реакции
Метод подстановки:
Заключается в подстановке экспериментальных данных ( , 1, 2,3; С0, С1, С2, С3) последовательно в уравнения кинетики реакции 0,1,2 и 3 порядков и определении, какое из них приводит к постоянному значению k. Порядок будет такой, какая формула дает лучшее постоянство k для различного времени.
Графический метод определения порядка реакции:
Заключается в построении графика выражающего зависимость различных функций С от . Чтобы определить порядок необходимо получить прямую линию функции. В случае реакции 1-го порядка линейную зависимость от дает lgC. В случае реакции 2-го порядка – 1/С от и для реакции 3-го порядка – 1/С2 от . Для реакции нулевого порядка С от дает параллельные прямые оси абсцисс.
Другой способ графического определения порядка реакции:
Порядок реакции определяется по зависимости скорости реакции от текущей концентрации реагирующего вещества L:
![]()



lg
,![]()

Lg
Lg
,a


 = lg k
= lg k
а
LgC1
LgC2
LgC



Рис.1.
Аналитические методы определения порядка реакции:
а) Порядок реакции определяется по начальной скорости реакции (метод Вант-Гоффа):
![]()
где 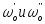 - начальные скорости реакции при начальных концентрациях
- начальные скорости реакции при начальных концентрациях .
.
б) Порядок реакции определяется по времени ’ и ’’ одинаковой степени превращения при начальных концентрациях Co’ и Co’’:
![]()
Лекция 3 влияние температуры на скорость химической реакции
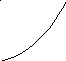
Температура сильно влияет на скорость химической реакции. Наблюдается несколько типов зависимости скорости реакции от температуры:


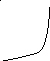
а
б
в


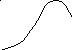
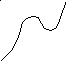

г![]()
е
д

T, C
а – нормальный характер зависимости, когда с увеличением температуры происходит быстрое возрастание скорости;
б – гетерогенные реакции в диффузионной области;
в – зависимость характерная для взрывчатых процессов (скачек скорости реакции при достижении температуры воспламенения);
г – каталитическая реакция, скорость которой определяется скоростью адсорбции (количество адсорбированного вещества уменьшается с возрастанием температуры);
д – характерная кривая для реакций осложненных побочными процессами, скорость которых становиться значительной при высоких температурах;
е – скорость реакции уменьшается с повышением температуры, например для O2+NO, при которой более полному равновесному превращению способствуют более низкие температуры.
Следует сказать, что зависимость типа а – характерна для простых реакций, другие же типы температурной зависимости – для сложных реакций или реакций, на протекание которых влияет скорость физических процессов.
Энергия активации
В 1884г. Вант-Гофф установил из равенства ![]() , чтоlnk должен быть линейной функцией от 1/Т. Это основано на том, что реакцию (1) можно рассматривать состоящей из двух уравнений: для прямой и обратной реакций, протекающих в равновесной системе. Результирующее уравнение имеет вид:
, чтоlnk должен быть линейной функцией от 1/Т. Это основано на том, что реакцию (1) можно рассматривать состоящей из двух уравнений: для прямой и обратной реакций, протекающих в равновесной системе. Результирующее уравнение имеет вид:
![]() ,
, ![]() (31)
(31)
Если принять, что ![]() иI- некая константа, то:
иI- некая константа, то:
![]() ,
, ![]() (32)
(32)
Если константа I= 0, то после интегрирования получим уравнение:
![]() ,
, ![]() (33 и 34)
(33 и 34)
или
![]() , (35)
, (35)
где ![]() K, K1 - константы интегрирования; А - постоянная величина.
K, K1 - константы интегрирования; А - постоянная величина.
В 1889г. Аррениус определил, что почти для всех реакций графики зависимости lnk от 1/T представляют собой прямые линии (см. Рис. 2). Впоследствии было выведено уравнение:
![]() ,
, ![]() (36 и 37)
(36 и 37)
где tg- A/R
При сравнении уравнений (35) и (36) очевидно, что E/RT=2,303a. Аррениус считал, что Е представляет собой разность энергий исходных реагентов и реагентов в активированном виде. Он предполагал также, что I=0, так как это соответствовало результатам его обработки для большинства известных тогда реакций. В 1909г. Величина Е была названа Траутцем энергией активации. Она является характеристикой реакции и определяет влияние температуры на скорость реакции. Из уравнения (35) видно, что константами характеризующими реакцию, являются предэкспонициальный множитель А и энергия активации Е. Когда реакцию проводят при двух различных температурах Т1и Т2 , то уравнение (35) можно записать в следующем виде: ![]() (38)
(38)



lnk

lnА

1/T (K)
Рис.2. График (зависимость) Аррениуса.
Параметр Е в случае простых реакций, протекающих в одну стадию показывает какой минимальной энергией, в расчете на 1 моль, должны обладать реагирующие молекулы, чтобы они могли вступить в реакцию.
В случае сложных реакций или реакций протекающих в несколько стадий Е не имеет строгого физического смысла и является некоторой функцией энергии активации отдельных стадий, обычно называемой эмпирической (кажущейся) энергией активации.
Никакой особой формы энергии, отвечающей энергии активации в молекулах нет. Энергия активации это то избыточное количество энергии (по сравнению со средней величиной), которой должна обладать молекула в момент столкновения, чтобы быть способной к химическому взаимодействию.
Избыточная энергия может быть:
-
Повышенная кинетическая энергия вращательного или поступательного движения.
-
Повышенная энергия взаимного колебания атомов или атомных групп в молекулах.
-
Повышенная энергия движения электронов.
Так, например, повышенная энергия движения электронов может достигаться при поглощении света (или от воздействия электромагнитных полей). Энергия электронов в атомах может повышаться при разрыве валентной связи (диссоциации, образование свободных радикалов или других атомов с ненасыщенной валентностью, при ударе молекул о стеку сосуда, электроразряд, ультразвук, излучения и т.д.). Обычно при рассмотрении вопросов химической кинетики приходиться иметь дело с системами, в которых доля активных молекул N/N = 10-10 – 10-20. Если меньше – скорость реакции чрезвычайно мала, если больше – реакция протекает со взрывом.
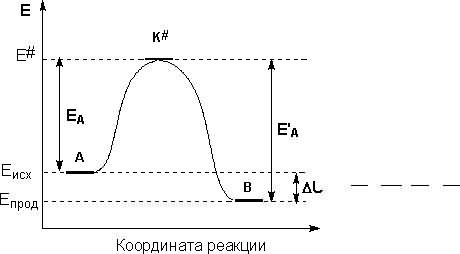
Интересно рассмотреть связь энергии активации с тепловым эффектом реакции. Химическую реакцию, протекающую при p= const, можно представить как переход системы из энергетического состояния 1 в состояние 2 с теплотой H. На рис. 3 видно, что переход системы из энергетического состояния 1 в состояние 2 возможен при затрате энергии Е1, а обратный переход - при затрате энергии Е2. При осуществлении реакции в прямом направлении выделяется энергия
![]() (39)
(39)
Величина -H равна разности энтальпий исходных и конечных продуктов реакции ![]() (40)
(40)
отсчитываемых от некоторого уровня, принятого за нулевой Н= 0.

StudFiles.ru
Формальная кинетика
Концентрационная цепь, составленная из электродов II рода: Ag, AgCl | KCl(a1) || KCl(a2) | AgCl, Ag
|
0 0 |
RT |
RT |
RT |
a1 |
|||||
|
Уравнение Нернста: E=Eпр −E лев=Eпр−E лев+ |
ln a1− |
ln a2= |
ln |
. |
|||||
|
F |
F |
F |
a2 |
||||||
Полярность определяется путем сравнения концентраций. Правым (положительным) будет тот электрод, который опущен в раствор с более высокой концентрацией хлорид-ионов,т. к. только в этом случае E > 0.
Скорость, константа скорости, основной постулат химической кинетики
Формальная кинетика рассматривает скорость химической реакции как функцию только концентрации.
Скорость любой гомогенной реакции по i-омукомпоненту определяется как изменение количества этого компонента в единицу времени в единице объема реакционного пространства. Для реакции aA + bB → cC + dD:
|
ri=− |
1 dnA |
=− |
1 |
dnB |
= |
1 |
dnC |
= |
1 |
dnD |
. |
||||||
|
V d τ |
V d τ |
V d τ |
V d τ |
||||||||||||||
Количество вещества в единице объема — это концентрация вещества:
|
r=− |
1 dCA |
=− |
1 dCB |
= |
1 dCC |
= |
1 dCD |
|||||||
|
a d τ |
b d τ |
c d τ |
d d τ |
|||||||||||
Скорость всегда больше нуля, поэтому при определении скорости через исходное вещество ставим знак «-»,т. к. концентрация исходных веществ убывает во времени.
Скорость зависит от концентраций исходных веществ, температуры, давления (в случае газофазной реакции).
Основной постулат химической кинетики — скорость реакции в каждый момент времени пропорциональна произведению концентраций исходных веществ, возведенных в некоторые
степени: r=k [ A]a [ B]b .
Коэффициент пропорциональности k называется константой скорости реакции. Константа скорости численно равна скорости химической реакции при единичной концентрации реагирующих веществ.
На размерность константы скорости влияет порядок реакции. Так для первого порядка k = [1/с], для второго порядка k = [л/моль*с]. Константа не зависит от концентрации реагирующих веществ, она постоянна при данной температуре.
Влияние температуры на константу скорости приближенно определяется правилом ВантГоффа: при повышении температуры на каждые 10 градусов скорость реакции увеличивается
|
k2 |
T 2−T1 |
||
|
в 2-4раза. |
=γ 10 , где k2 – константа при новой температуре, k1 – константа при |
||
|
k1 |
|||
исходной температуре, γ — коэффициент Вант-Гоффа.
Кинетические кривые
Кинетической кривой называется график зависимости концентрации i-гокомпонента

реакции от времени.
Найдем зависимость концентрации от времени для реакций 0-3порядков. Скорость реакции нулевого порядка не изменяется с течением времени:
|
−dCA |
=k dC A=−kdt C A=−kt+C0 - линейная убывающая функция для исходного |
||||||||
|
dt |
|||||||||
|
компонента. C0 – концентрация компонента A в начале реакции. |
|||||||||
|
Реакция первого порядка: |
−dCA |
=k [A] |
dC A |
=−kdt lnCA=−kt+lnC0 |
C A=C0 e |
−kt |
- для |
||
|
dt |
[ A] |
||||||||
исходного компонента А график — убывающая экспонента.
Реакция второго порядка, если концентрации исходных веществ равны:
|
−dCA |
2 |
dC A |
1 |
1 |
C0 |
||||||||||||
|
=k [A] |
=−kdt− |
=−kt− |
C A= |
- убывающая гипербола. |
|||||||||||||
|
dt |
[ A]2 |
C A |
C 0 |
C0 kt+1 |
|||||||||||||
|
Реакция третьего порядка, если концентрации исходных веществ равны: |
|||||||||||||||||
|
−dCA |
dC A |
||||||||||||||||
|
3 |
1 |
1 |
1 |
||||||||||||||
|
=k [A] |
=−kdt− |
=−kt− |
C A= |
||||||||||||||
|
dt |
3 |
2 |
2 |
1 |
|||||||||||||
|
[ A] |
C A |
C0 |
√kt+ |
||||||||||||||
|
C02 |
|||||||||||||||||
Тангенс угла наклона касательной, проведенной к какой-либоточке кривой, будет равен скорости реакции в данный момент времени по данному компоненту.
Для наглядности кинетические кривые строят в таких координатах, в которых кинетическая кривая становится прямой, т. е. линеаризуется. Для реакции 1 порядка это координаты lnC=f(t), для 2 порядка — 1/C = f(t).
Порядок реакции r=k [A]a [B]b
Частным порядком реакции называют порядок реакции по i-мукомпоненту. a – порядок реакции по компоненту a, b – порядок реакции по компоненту b. Общий порядок реакции — это сумма частных порядков: n = a + b.
Молекулярностью реакции называется количество частиц, принимающих участие в одном элементарном акте химического превращения.
Различают моно-,би- и тримолекулярные реакции. Участие в реакции более трех частиц следует рассматривать как сложную реакцию, протекающую через несколько элементарных стадий, каждая из которыхмоно-,биили тримолекулярна. Для простых одностадийных гомогенных реакций молекулярность совпадает с порядком реакции.
Временем полупревращения называется такой промежуток времени, за который исходная концентрация реагентов снизится вдвое.
Найдем время полупревращения для реакций 0-3порядков.
Реакция нулевого порядка: C A=−kt+C 0 C20 =−kt1/ 2+C0 t1 /2= C2k0 Реакция первого порядка:C A=C0 e−kt C20 =C0 e−kt1/2 ln12 =−kt1 /2 t1/ 2= ln2k

|
Реакция второго порядка: |
C |
= |
C0 |
C0 |
= |
C0 |
C |
kt |
+1=2t |
= |
1 |
||||||||||||||||
|
C0 kt+1 |
2 |
C |
0 kt1/ 2+1 |
kC0 |
|||||||||||||||||||||||
|
A |
0 |
1/ 2 |
1/ 2 |
||||||||||||||||||||||||
|
1 |
1 |
1 |
1 |
3 |
|||||||||||||||||||||||
|
Реакция третьего порядка: |
C A=√ |
k= |
( |
− |
) t1/ 2= |
||||||||||||||||||||||
|
kt+ |
1 |
2t |
C 2 |
C02 |
2kC02 |
||||||||||||||||||||||
|
C02 |
|||||||||||||||||||||||||||
Степень превращения — это отношение количества молей вещества, вступившего в реакцию,
|
к исходному числу молей: α |
= |
n |
= |
C0 −C |
=1− |
C |
. |
|
|
ni |
C0 |
C 0 |
||||||
|
i |
Интегральные методы нахождения порядка реакции.
1)Метод подстановки — подставляем полученные значения времени и концентрации исходного вещества в кинетические уравнения разных порядков, находим константы реакции. Порядок реакции тот, в каком уравнении константа остается постоянной для всех значений.
2)Графический метод — строим графики в координатах C – t, lnC – t, 1/C – t, 1/C2 – t. Какой из этих графиков окажется прямой линией, такой порядок у реакции.
3)Способ определения времени полупревращения — проводим несколько опытов при различных начальных концентрациях и определяем периоды полупревращения. По зависимости периода полупревращения от начальной концентрации определяем порядок реакции. В рамках этого способа можно вычислить порядок реакции по этой
формуле: n= lg t'1'/ 2−lg t'1/ 2 +1 , зная периоды полупревращения при двух различныхlg c''0 −lg c'0
начальных концентрациях. Порядок реакции может быть дробным.
4)Метод Эмануэля-Кноррепозволяет вычислить порядок реакции по формуле:
|
ln[ |
t ' |
] |
|||||||
|
n=1+ |
(t ' '− |
t ') |
, где α '=1− |
c' |
- степень превращения вещества в момент |
||||
|
lg (1−α ') |
c0 |
||||||||
времени t', t' – произвольно выбранный момент времени, t'' – такой момент времени, в который c'' = (c')2.
Дифференциальные методы определения порядка реакции.
Реакция aA + bB → продукты.
|
1 dcA |
nA |
nB |
|||||
|
Скорость реакции r=− |
=k1 cA |
cB |
, где nA, nB – частные порядки реакций. |
||||
|
a dt |
|||||||
Метод изоляции Оствальда. Проводим нужную реакцию сначала в условиях избытка всех исходных веществ, кроме одного. Тогда можно пренебречь изменением концентраций всех реагентов, кроме того, по которому ведется расчет. Тогда концентрацию всех компонентов можно внести в постоянный коэффициент, и скорость реакции можно записать так:
|
−rA=− |
dc1 |
=k1 c1n1 , где |
k1=akc20n2 |
, n1 – порядок реакции по первому веществу. |
|
dt |
||||
|
Способ логарифмирования |
||||
|
Прологарифмируем это выражение: |
ln−rA=lnk 1+n1 lnc1 . |
|||
Построим график в координатах ln(-rA)– lnc1, все точки должны лечь на прямую линию.
Тангенс угла наклона этой прямой будет равен порядку реакции по веществу А. Этим методом определяется концентрационный порядок.
Влияние температуры на скорость химической реакции
Влияние температуры на константу скорости приближенно определяется правилом ВантГоффа: при повышении температуры на каждые 10 градусов скорость элементарной
|
k2 |
T 2−T1 |
||
|
гомогенной реакции увеличивается в 2-4раза. |
=γ 10 , где k2 – константа при новой |
||
|
k1 |
|||
температуре, k1 – константа при исходной температуре, γ — коэффициентВант-Гоффа.КоэффициентВант-Гоффаявляется функцией температуры, при очень высоких и очень низких температурах он становится равным единице, т. е. реакция перестает зависеть от изменения Т.
Более полно зависимость скорости реакции от температуры описывает уравнение Аррениуса:
|
дlnk |
= |
Ea |
, где Ea – энергия активации — минимальная избыточная энергия по |
|
|
дT |
RT 2 |
|||
сравнению со средней, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
|
Ea |
; k= Ae− |
Ea |
|||
|
Проинтегрировав это уравнение, получаем: lnk=lnA− |
RT |
. A – |
|||
|
RT |
|||||
предэкспоненциальный множитель — численно равен константе скорости химической реакции при бесконечно большой температуре.
Построив график в координатах lnk – 1/T, получим прямую линию, которая отсекает на вертикальной оси отрезок длиной lnA, а тангенс угла наклона которой равен Ea/R.
Для расчета энергии активации необходимо знать значений констант скоростей при различных температурах, чтобы построить данный график.
Уравнение Аррениуса позволяет рассчитать константу скорости при определенной температуре, зная константу скорости при другой температуре:
ln kk21= ERa (T11− T12)
Для определения энергии активации в первом приближении достаточно знать две константы скорости при двух различных Т.
Температурный коэффициент Вант-Гоффаможно связать с энергией активации:
|
k |
2 |
Ea |
( |
1 |
1 |
) |
k |
2 |
=exp[ |
Ea |
( |
1 |
1 |
)]=γ |
T 2−T1 |
||||||||||||||||||||||||
|
ln |
= |
− |
− |
10 |
|||||||||||||||||||||||||||||||||||
|
k |
1 |
R |
T 1 |
T 2 |
k |
1 |
R |
T 1 |
T 2 |
||||||||||||||||||||||||||||||
|
E T −T |
10 |
E T −T |
10 |
E |
10 |
||||||||||||||||||||||||||||||||||
|
T |
−T |
||||||||||||||||||||||||||||||||||||||
|
γ =exp[ |
a |
2 1 |
] |
2 |
1 =exp[ |
a |
2 1 |
]=exp[ |
a |
] |
|||||||||||||||||||||||||||||
|
R |
T 1 T2 |
R |
T 1 T2 |
T 2−T1 |
R |
T 1 T2 |
|||||||||||||||||||||||||||||||||
|
lnγ = |
10 Ea |
Ea =0,1 R T1 T2 ln γ |
|||||||||||||||||||||||||||||||||||||
|
RT 1 T2 |
|||||||||||||||||||||||||||||||||||||||
StudFiles.ru
Читайте также
 Методы определения основного обмена
Методы определения основного обмена Метод обучения это в педагогике определение
Метод обучения это в педагогике определение Базисно индексный метод определения сметной стоимости
Базисно индексный метод определения сметной стоимости Определение твердости методом бринелля
Определение твердости методом бринелля Методы определения нмцк
Методы определения нмцк Методы определения твердости
Методы определения твердости Методы определения предполагаемой массы плода
Методы определения предполагаемой массы плода- Методы определения качества продукции
- Методы определения избыточной численности
 Методы определения длины корневого канала
Методы определения длины корневого канала Методы определения биологического возраста
Методы определения биологического возраста Методы определения таможенной стоимости
Методы определения таможенной стоимости
 Методы определения основного обмена
Методы определения основного обмена Метод обучения это в педагогике определение
Метод обучения это в педагогике определение Базисно индексный метод определения сметной стоимости
Базисно индексный метод определения сметной стоимости Определение твердости методом бринелля
Определение твердости методом бринелля Методы определения нмцк
Методы определения нмцк Методы определения твердости
Методы определения твердости Методы определения предполагаемой массы плода
Методы определения предполагаемой массы плода Методы определения длины корневого канала
Методы определения длины корневого канала Методы определения биологического возраста
Методы определения биологического возраста Методы определения таможенной стоимости
Методы определения таможенной стоимости